CAR-T-Related Toxicity
Lenzilumab, a novel, first-in-class monoclonal antibody, binds to and neutralizes circulating GM-CSF. Lenzilumab is a potent inhibitor of GM-CSF in vivo and also has shown to be safe and tolerable in 100 patients over several previous clinical trials. Strong scientific rationale is emerging to point to GM-CSF as the essential driver of a harmful inflammatory cascade behind CAR-T-induced neurotoxicity (NT) and cytokine release syndrome (CRS). These side effects limit both the number of patients that can be treated with CAR-T and the dosage that can be safely administered. They also require intensive hospital monitoring of CAR-T patients during therapy.
Lenzilumab could aid in the expansion of CAR-T by allowing for a higher CAR-T dose, potentially improving efficacy and reducing patient hospitalization. Hospitals now must ensure that a place in the ICU is available for each CAR-T patient prior to treatment, limiting the potential for true outpatient administration and adding significant costs and patient management hurdles to the therapy. Current treatment of these side effects mostly revolves around tocilizumab, an IL6 receptor antagonist, which helps with CRS, but is not effective with neurotoxicity. An anti-GM-CSF approach with lenzilumab could prevent neurotoxicity and CRS, a potential key differentiator in clinical practice. Lenzilumab is poised to not only improve the clinical management and patient outcomes of CAR-T cell therapy, but also significantly reduce hospitalization costs, while enabling the treatment of more patients.
Humanigen has pre-clinical animal model work underway for lenzilumab in CAR-T toxicities. Leading key opinion leaders are designing studies to explore lenzilumab as a prophylactic treatment to minimize neurotoxicity associated with CAR-T. Overall, lenzilumab has the potential to make CAR-T safer, better and used more routinely and widely.
Click here for a Humanigen report on the science behind how lenzilumab could play a key role in CAR-T therapy.
Potential to improve CAR-T
Immunotherapies are a new generation of cancer treatments, poised to radically change clinical oncology with their specificity and high response rates. CAR-T therapy, a new form of immunotherapy, has been approved twice by the FDA to date with over 300 clinical trials currently in process. High remission rates are being observed in cancers unresponsive to radiation and chemotherapy. However, CAR-T cell therapy is currently limited by the risk of life-threatening neurologic toxicity (NT) and CRS. Despite active management, all CAR-T responders experience some degree of CRS. Up to 50% of patients treated with CD19 CAR-T have at least Grade 3 CRS or neurotoxicity. CRS and neurotoxicity are limiting the number of patients treated by CAR-T therapy and the dosage administered.
Reducing or eliminating CRS and neurotoxicity in these therapies would provide great value. Determining what is driving or exacerbating the signature CAR-T inflammatory response is a crucial first step. Although a plethora of cytokines, signaling molecules, and cell types are involved in this pathway, one cytokine appears to be at the heart of CRS and neurotoxicity, GM-CSF. Normally undetectable in human serum, it is central to the cyclical positive feedback loop that drives inflammation to the extremes of cytokine storms and endothelial activation. Neurotoxicity and cytokine storms are not the result of a simultaneous release of cytokines, but rather a signaling cascade of inflammation initiated by GM-CSF that results in the trafficking and recruitment of myeloid cells to the tumor site. These myeloid cells produce the cytokines observed in CRS and NT, perpetuating the inflammatory cascade.
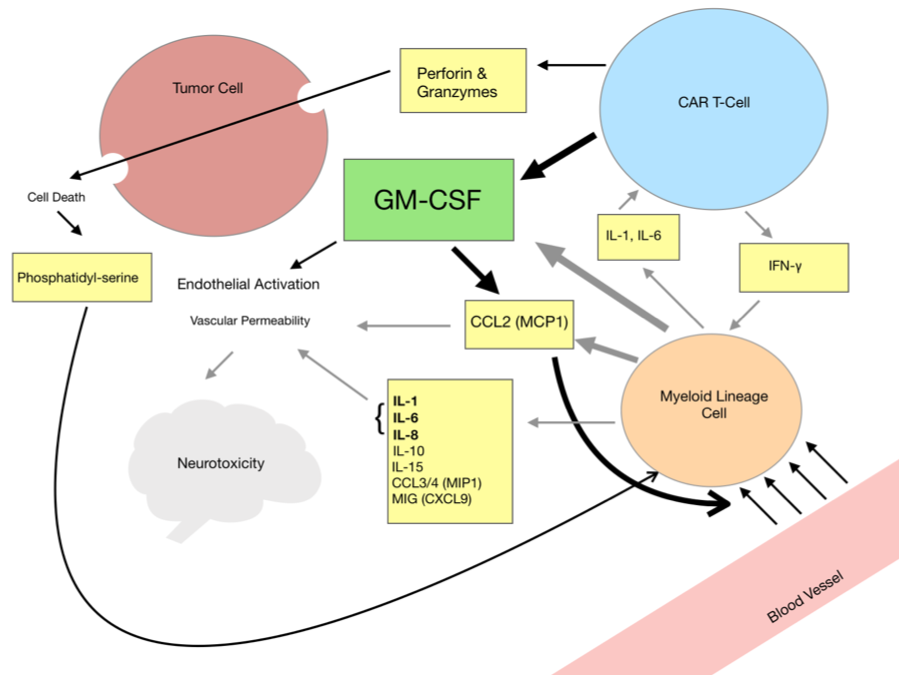
Neurotoxicity and CRS: GM-CSF Initiates the Cascade
The mechanism by which GM-CSF initiates the inflammatory cascade is becoming clear. A key component of the cascade is its ability to self perpetuate, a phenonemon for which the CCR2+ myeloid cells are largely responsible. GM-CSF is the driver causing CCR2+ myeloid cells to traffic to the site of inflammation and initiating a self-perpetuating inflammatory cascade within tissues, including the central nervous system (CNS)1. The CNS is particularly vulnerable to monocyte-derived phagocytes and GM-CSF drives the activation, trafficking, and recruitment of these myeloid cells2. CCR2+ myeloid cells produce CCL2 (MCP1) and possess a receptor for CCL2, a critical chemokine in signaling a cascade. Thus, CCL2 positively mediates its own production in a self-reinforcing manner. After recruitment to the tumor site, myeloid cells produce IL-1 and IL-6 which induce further GM-CSF production by CAR-T3. GM-CSF stimulates cytokine production from myeloid cells, resulting in a positive and cyclical signaling pathway of inflammation. Preventing myeloid cell recruitment by GM-CSF is crucial in preventing CRS and neurotoxicity. It has been demonstrated that tumor sites of GM-CSF knockout CARs produce less CCL2 (MCP1), CCL5, and CCL3 (MIP1)4. These are key chemokines for both trafficking and recruitment of myeloid cells.
1. Croxford, A. et. al (2015). The Cytokine GM-CSF Drives the Inflammatory Signature of CCR2+ Monocytes and Licenses Autoimmunity. Immunity, 43(3), 502-514. 2. Spath, S. et. al (2017). Dysregulation of the Cytokine GM-CSF Induces Spontaneous Phagocyte Invasion and Immunopathology in the Central Nervous System. Immunity, 46(2), 245-260. 3. Barret, D. et. al (2016). Interleukin 6 Is Not Made By Chimeric Antigen Receptor T Cells and Does Not Impact Their Function. Blood. 4. Sentman, M.-L., et. al (2016). Mechanisms of Acute Toxicity in NKG2D Chimeric Antigen Receptor T Cell-Treated Mice. The Journal of Immunology, 197(12), 4674-4685
Please see the lenzilumab References section for a compendium of the current body of evidence supporting the scientific rationale to target GM-CSF to improve CAR-T therapy.
CMML
Phase I Lenzilumab Study Underway in CMML Patients
GM-CSF also stimulates white blood cell production and is the hallmark of some hematologic malignancies – such as chronic myelomonocytic leukemia (CMML) and juvenile myelomonocytic leukemia (JMML) – and perhaps some solid tumors. In addition, it is tied to inflammation in several significant, chronic autoimmune diseases.
GM-CSF is one of the cytokines involved in the development and maturation of certain types of myeloid blood cells. Earlier data generated by a collaborator confirmed that hypersensitivity to GM-CSF plays an important role in the growth and survival of CMML cells. Such hypersensitivity is also a hallmark of JMML. Inhibition of GM-CSF may affect growth of leukemic cells in patients. CMML is an orphan disease and JMML is a rare pediatric disease.
Humanigen continues to study lenzilumab as a potential treatment for CMML and JMML. A Phase I trial focused on patients with previously treated CMML is underway. The Phase I study is a multi-center, open-label dose escalation trial to evaluate the safety, maximum tolerated dose and preliminary activity of single-agent lenzilumab in CMML patients who are relapsed, refractory to, or intolerant to standard-of-care treatments. The first patient was dosed with lenzilumab in late July 2016.
The study will enroll up to 18 patients and is designed to identify the maximum tolerated dose -and a recommended Phase II dose – of lenzilumab in previously treated CMML patients. It will also assess possible preliminary efficacy of single-agent lenzilumab and provide additional data on pharmacokinetics, pharmacodynamics – including correlative biomarkers and mutational analysis – and safety. This data could be used to begin a study in JMML.
Currently available treatments in CMML are limited and produce inadequate responses, leaving these patients with high unmet medical need for effective and tolerable therapies.